If you spend any time working on a laptop or using a smart phone, you’ve noticed that computers are lighter, smaller and more powerful than ever. But battery capacities have had a tough time keeping pace with the added drain. Much of that battery lag stems from a lack of new energy-storing materials.
The challenge of building a better battery illustrates a fundamental bottleneck in developing new materials for a variety of applications, including hydrogen storage, fuel cells and solar cells. The traditional model for any new chemistry has involved researchers’ hatching an educated guess, synthesizing compounds in a laboratory, then testing them to see if they work.
But even under the best circumstances, developing a new material for an application and optimizing it for commercial use typically takes two decades, says Anubhav Jain, a graduate student in Gerbrand Ceder’s group at the Massachusetts Institute of Technology. A Department of Energy Computational Science Graduate Fellowship supports Jain’s work.
Earlier this summer, at the SciDAC 2010 meeting in Chattanooga, Tenn., Jain presented the MIT researchers’ progress in what they’re calling the Materials Genome Project. It’s a computational encyclopedia that will allow chemists and materials scientists to reduce the guesswork by focusing experiments on compounds that show the most promising properties.
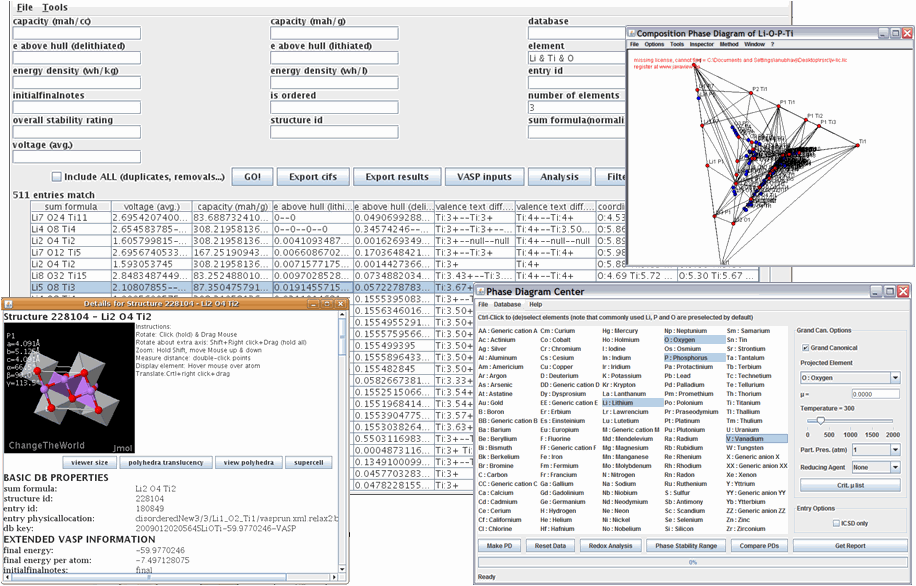
Custom software to explore data generated by high-throughput materials computation allows scientists to search for materials satisfying certain constraints, display details on materials and find trends and features in the data. For example, the program can determine computational phase diagrams for any chemical system, allowing researchers to better understand thermodynamic stability or find new materials.
Scientists have reported developing nearly 100,000 new inorganic compounds – but have done little to tease out their potentially useful properties. The Materials Genome Project could help fill those information gaps. It has a two-pronged approach: The researchers use computational data to estimate properties, which are compiled in a Web database. And they predict the behavior of materials yet to be synthesized.
High-throughput computing allows Ceder’s group to evaluate materials quickly. Like the methods medicinal chemists use to screen for hundreds of drug candidates, the MIT researchers run approximately 200 calculations at the same time.
Advances in computing power and computational chemistry over the past 20 years have made this approach possible. Computational chemistry requires solving known physics equations that predict the behavior of materials. A basis for those calculations is density functional theory (DFT), a systematic framework that breaks a material down into identical repeating components, or cells, in a defined geometric arrangement.
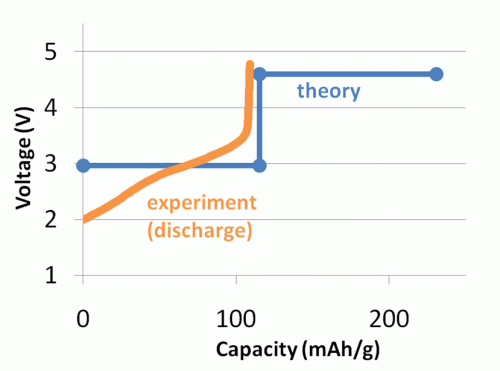
A graphic comparison of computed and experimental voltage profiles for a proposed battery material. Click image to enlarge and for more information.
The number of electrons in the cell determines a calculation’s complexity. A single simple material with a cell that contains two atoms could be done easily on a desktop computer. Complex structures require much larger cell sizes – up to 1,000 atoms, complexity that brushes computational limits.
By definition, crystalline materials have an ordered, repeating atomic structure, so DFT works well for these compounds, which typically include between 1 and 200 atoms. DFT considers the interactions inside each cell, simultaneously knitting the cells to form a picture of the entire material, much like the repeating tessellations in an Escher drawing.
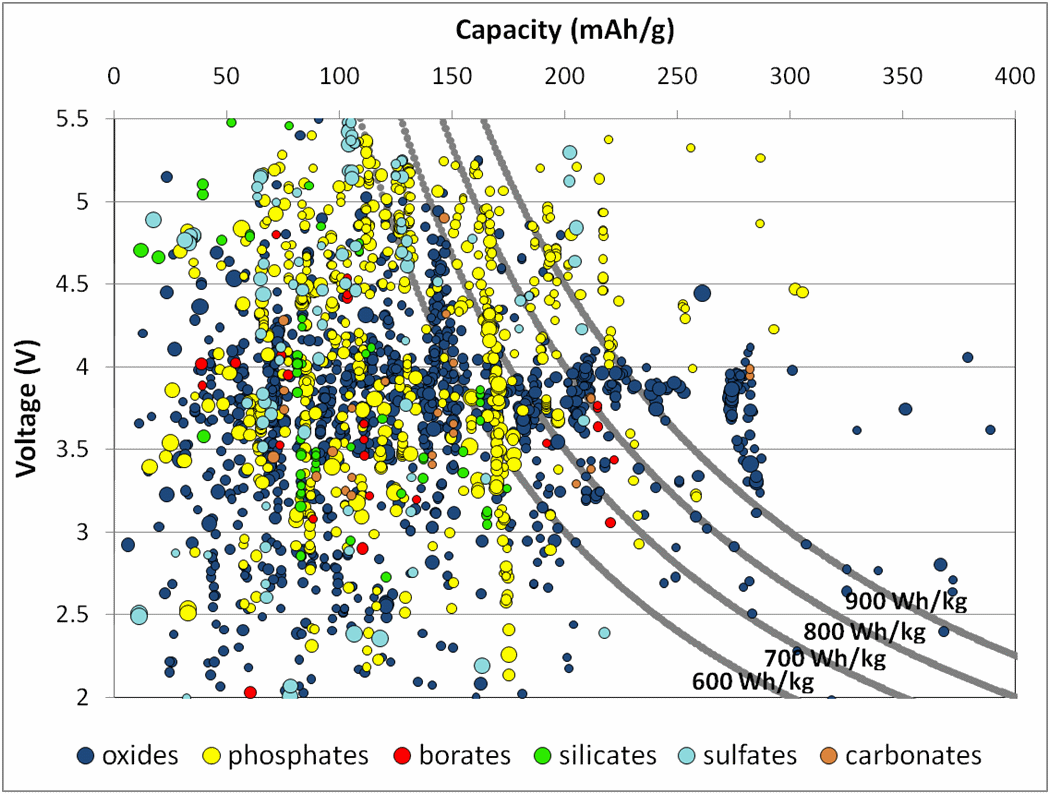
The product of voltage and capacity provides the gravimetric energy density – how much energy a cathode material could theoretically store per unit mass. (For this graph, the units for capacity are milliampere-hours per gram, or mAh/g.) Current cathode materials typically achieve energy densities in the 600-700 Watt-hours per kilogram (Wh/kg) range, shown by the gray lines. Many materials are theoretically capable of 700Wh/kg, better than current materials on the market. The plot hints that different chemistries operate under different voltage ranges (represented by the different colors). A commercial battery must be under 4.5V to be compatible with the electrolyte, largely restricting certain chemistries like sulfates but hardly affecting others like oxides and borates.
The Materials Genome is possible because DFT calculations are now accurate enough to predict material properties, rather than just confirming experimental values. For certain types of calculations, such as voltage and other battery-related properties, DFT calculations match laboratory-measured values. For example, with a lithium ion battery, DFT voltage calculations fell within 3 percent of the values measured in experiments.
For other calculations, DFT is less accurate but still provides useful information. A property critical to the viability of solar cells – the band gap – is one such case. In this context, the band gap is the minimum energy of light that a material can absorb. DFT might not offer reliable values for the band gap, but the calculations can help researchers compare and rank candidate materials – for example, if the band gap value for material A is higher than that for material B.
Voltage and band gap are just a couple of basic properties for which understanding is often spotty because they might not have been important to researchers when they synthesized the materials.
Researchers substituted different chemical combinations to produce 20,000 new compounds.
For example, magnesium diboride was discovered half a century ago, but only this past decade have researchers tried it as a high-temperature superconductor. Lithium iron phosphate was discovered more than 70 years ago, but only over the past 15 have researchers investigated its battery properties. Because useful materials probably are sitting on laboratory shelves, DFT calculations provide an efficient way to add missing information to this large catalog of compounds.
The Ceder group is nearly 80 percent finished with the Materials Genome. Jain and his colleagues are already demonstrating how this information and the methods used to collect it could be applied for further materials discovery – for capturing carbon, for thermoelectric materials that convert waste heat to electricity and a range of other materials applications.
Batteries have provided the Ceder group a way to demonstrate the power of this computational-discovery approach. In a step beyond cataloging materials, the researchers also have used calculations to look for promising undiscovered materials.
They started their search with 1,000 compounds that contained lithium and a transition metal, a common battery chemical combination, and uncovered a potential energy-storage compound. Then they took their computations one step further: Using these materials as a template, the researchers substituted different chemical combinations to produce 20,000 new compounds. With DFT, they calculated the total energy of these hypothetical materials.
Based on their calculations, Ceder and colleagues could eliminate high-energy candidates. Such compounds are unlikely to be chemically stable, which is a prerequisite for their production in the laboratory. With the narrower field of lower energy materials, Ceder’s team could do further calculations with suitable candidates to look for good battery properties.
The Ceder group has synthesized the three most promising materials from this search – a previously known material and two others that have not previously been synthesized. One is a new class of cathode materials, lithium metal carbonophosphates, Li3M(CO3)(PO4). They’ve also run initial electrochemical tests to look at the battery materials’ properties.
Optimizing these properties is up next. By changing material particle sizes or adding coatings to improve the movement of electrons, researchers hope to improve the materials’ experimental properties.
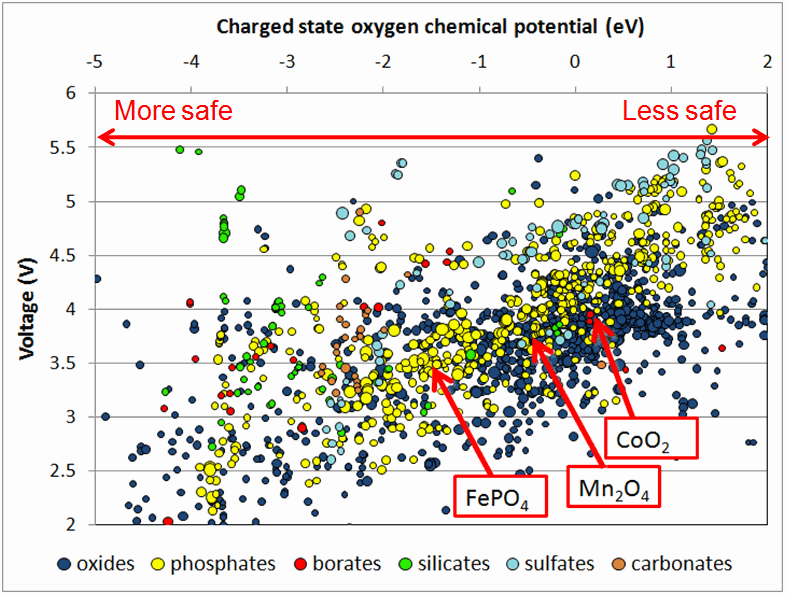
Besides energy-storage qualities, battery materials must be chemically stable, particularly when subjected to conditions outside a laboratory. In March in the journal Electrochemistry Communications, Ceder’s group reported using Materials Genome data to compare the safety of two possible cathode materials: lithium iron phosphate and lithium manganese phosphate. Although the manganese compound has higher voltage than the iron compound, calculations demonstrate that it also has a lower thermal stability. Studying these effects computationally before making the compounds in the laboratory could save months of valuable experimental time. He is now comparing voltage and safety across thousands of compounds and finding that high-voltage materials are generally less safe. However, he adds, certain chemistries may offer the possibility of both high voltage and high safety.
Jain and his colleagues are already looking at other applications for this research, including ways materials can sponge up pollutants from coal gasification. Although gasification improves the thermal efficiency of fossil fuel burning, the process produces mercury and other toxic chemicals. In research reported in May in the journal Chemical Engineering Science, the researchers used DFT calculations to seek metals that can adsorb mercury. Palladium was most effective, followed by platinum, a finding consistent with experiments.
Jain and colleagues also are looking at DFT’s reliability for identifying solar cell materials. Compared with batteries, solar cells present additional computational challenges for the Ceder group’s high-throughput approach. DFT does well at predicting the chemistry of materials in their ground state, without added energy input. But to understand the excited energy states necessary for new solar materials, researchers must approximate materials’ ability to absorb energy. Other more computationally demanding techniques are available, but they might not be necessary. Jain wants to learn how close he can come using DFT predictions.
The MIT team thinks it’s worth a try. Where it has taken traditional methods two decades to discover roughly a dozen potential new battery cathode materials, Ceder’s group has come up with three more in just four years.
“When we put this data online, people will find additional design opportunities,” Jain says, such as new transparent conducting oxides for touch screens or other related technologies.
Adds Jain, “Researchers around the world will apply the data in ways we can’t even foresee right now, greatly accelerating the pace of discovery across many different fields.”